- Akesson LS, Savarirayan R. Fibrodysplasia Ossificans Progressiva. 2020 Jun 11 [Updated 2024 May 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK558090
- Kaplan FS, Shore EM, Pignolo RJ. Fibrodysplasia ossificans progressiva emerges from obscurity. Trends Mol Med. 2025 Feb;31(2):106-116. doi: 10.1016/j.molmed.2024.08.010. Epub 2024 Sep 18. PMID: 39299836.
- Pignolo RJ, Baujat G, Brown MA, De Cunto C, Hsiao EC, Keen R, Al Mukaddam M, Le Quan Sang KH, Wilson A, Marino R, Strahs A, Kaplan FS. The natural history of fibrodysplasia ossificans progressiva: A prospective, global 36-month study. Genet Med. 2022 Dec;24(12):2422-2433. doi: 10.1016/j.gim.2022.08.013. Epub 2022 Sep 24. PMID: 36152026.
- Bildnachweis: Fred Kaplan, Mütter Museum, College of Physicians of Philadelphia - Shafritz et al., 1996
- Bildnachweis: Alabama medicine: journal of the Medical Association of the State of Alabama 1984
Fibrodysplasia ossificans progressiva
FOP ist eine seltene genetische Erkrankung, bei der sich Weichgewebe in Knochen verwandelt, was die Bewegungsfreiheit einschränkt und schwere Komplikationen verursacht, mit einer durchschnittlichen Lebenserwartung von 40 Jahren.
FOP: die „Steinmann-Krankheit“
Fibrodysplasia ossificans progressiva (FOP), auch als „Steinmann-Krankheit“ bekannt, ist eine seltene autosomal-dominante genetische Erkrankung, die den Bewegungsapparat und das Bindegewebe betrifft. Laut Veröffentlichungen variiert die Prävalenz zwischen 0,36 und 1,36 pro Million Einwohner, ohne Berücksichtigung ethnischer oder geografischer Faktoren.
Kinder mit FOP erscheinen bei der Geburt normal, mit Ausnahme von angeborenen Fehlbildungen der großen Zehen (Hallux valgus, Fehlbildung des ersten Mittelfußknochens und/oder Monofalangeus). Im ersten Lebensjahrzehnt können sporadisch Episoden mit schmerzhaften Weichteilschwellungen auftreten, die häufig auf Verletzungen, intramuskuläre Injektionen, Virusinfektionen, Muskelzerrungen, Stürze oder Erschöpfung zurückzuführen sind. Diese Schübe verwandeln Skelettmuskeln, Sehnen, Bänder, Faszien und Aponeurosen in heterotope Knochen, wodurch Bewegungen unmöglich werden. Es wurden Patienten mit atypischen Formen von FOP beschrieben, die die klassischen Anzeichen von FOP in Verbindung mit einem zusätzlichen Anzeichen oder mehreren atypischen Anzeichen (gleichzeitige aplastische Anämie, Kraniopharyngeom, Glaukom im Kindesalter, Wachstumsverzögerung; FOP plus) oder erhebliche Abweichungen bei einem oder beiden klassischen Anzeichen von FOP (normale Zehen oder starke Verkürzung der Finger; FOP-Varianten) aufweisen.
FOP steht in Zusammenhang mit pathogenen Varianten des ACVR1-Gens (Activin-Typ-1-Rezeptor, auch bekannt als ALK2), das den Activin-A-Typ-1-Rezeptor für verschiedene Liganden, darunter BMP (Bone Morphogenetic Protein), kodiert. Die Diagnose wird durch klinische und radiologische Befunde nahegelegt und später durch die molekulare Untersuchung bestätigt, die im Rahmen einer genetischen Beratung durchgeführt wird. Die Differentialdiagnose umfasst progressive Knochenheteroplasie, Osteosarkom, Lymphödem, Weichteilsarkom, Desmoidtumore, juvenile aggressive Fibromatose und nicht erbliche heterotope Ossifikation (HO).
Die Prognose von POF bleibt aufgrund der schweren Morbidität, der progressiven funktionellen Progression und der frühen Mortalität ungünstig. Der Verlauf ist durch das Auftreten einer besonders behindernden axialen und peripheren Ankylose gekennzeichnet, die dazu führt, dass die Betroffenen ab Beginn des dritten Lebensjahrzehnts bei alltäglichen Aktivitäten auf die Hilfe von einer oder zwei Personen angewiesen sind.
Die wichtigsten Komplikationen sind kardiopulmonale (wiederholte Pneumopathie, Rechtsherzversagen, restriktive Ateminsuffizienz), neurologische (Kopfschmerzen, neuropathische Schmerzen, Allodynie, Myoklonus), renale (Lithiasis), kardiale (Überleitungsstörungen), kraniofaziale und otorhinolaryngologische (TMJ-Dysfunktion, Schluckstörungen, Taubheit).
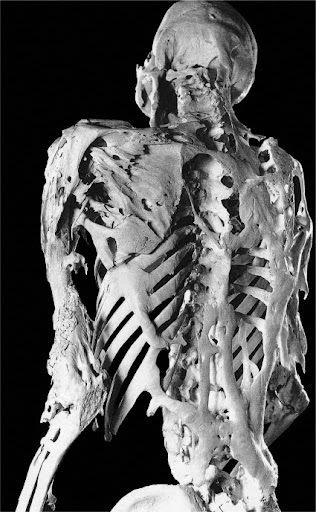
Abbildung 1: Fortgeschrittene knöcherne Manifestationen von FOP bei einem 39-jährigen Mann.4
Derzeit gibt es keine definitive Behandlung. Die Anwendung von hochdosierten Kortikosteroiden über einen Zeitraum von vier Tagen, beginnend innerhalb der ersten 24 Stunden nach dem Aufflammen, kann die akute Entzündung und das Gewebeödem, die in den frühen Stadien der Krankheit beobachtet werden, verringern. Die Prävention basiert auf der Umsetzung von Maßnahmen zur Reduzierung von Stürzen (verbesserte Sicherheit zu Hause, Verwendung eines Schutzhelms), zur Verhinderung einer Verschlechterung der Atemwege (Anreiz-Spirometrie) und von Virusinfektionen. Die durchschnittliche Lebenserwartung beträgt etwa 40 Jahre.
Die natürliche Entwicklung von FOP: eine prospektive, globale 36-Monats-Studie
Im Jahr 2022 zeigte eine von Ipsen durchgeführte globale Studie zur natürlichen Entwicklung von FOP die lähmenden Auswirkungen der Krankheit auf das Leben der Patienten. Diese prospektive, internationale Studie verfolgte 114 Personen mit klassischer FOP (ACVR1R206H-Mutation) über einen Zeitraum von 36 Monaten, um das Fortschreiten der Krankheit unter Standard-Versorgungsbedingungen zu untersuchen.
Wichtige klinische Erkenntnisse
- Schübe und HO-Entwicklung: 71,9 % der Teilnehmer erlitten Schübe, vorwiegend im oberen Rücken (17,9 %), in der Hüfte (14,8 %) und in der Schulter (10,9 %). Die Bildgebung zeigte, dass 26,9 % der Schübe innerhalb von 84 Tagen zur Bildung neuer HO führten.
- HO-Belastung: Die Ganzkörper-Computertomographie (WBCT) zeigte, dass das mittlere HO-Ausgangsvolumen 314,4 × 10³ mm³ betrug und mit zunehmendem Alter anstieg. Die höchste Rate an HO-Bildung wurde bei Jugendlichen und jungen Erwachsenen beobachtet (41,5 × 10³ mm³/Jahr in der Gruppe der 15- bis 25-Jährigen).
- Funktioneller Rückgang: Es wurde eine fortschreitende Gelenkverengung beobachtet, wobei viele Personen bereits mit 20 Jahren auf den Rollstuhl angewiesen waren. Die Werte der Cumulative Analogue Joint Involvement Scale (CAJIS) stiegen über einen Zeitraum von drei Jahren leicht an, aber der Einsatz von Hilfsmitteln nahm deutlich zu.
- Atemwegskomplikationen: Thoraxinsuffizienz, eine der Hauptursachen für Morbidität, lag bei 5,3 % zu Studienbeginn vor und verschlechterte sich im Laufe der Zeit. Die forcierte Vitalkapazität (FVC) nahm mit zunehmendem Alter ab und stabilisierte sich nach der Adoleszenz bei unter 50 % der prognostizierten Werte.
- Skelett- und Wachstumsanomalien: Alle Patienten wiesen Fehlbildungen der großen Zehe auf; 34,5 % hatten Hüftkopfanomalien und 14 % eine Verkürzung des Schenkelhalses. Bei pädiatrischen Patienten wurden dichte metaphysäre Bänder (DMBs) festgestellt, die wahrscheinlich mit den Auswirkungen der chronischen Erkrankung zusammenhängen.
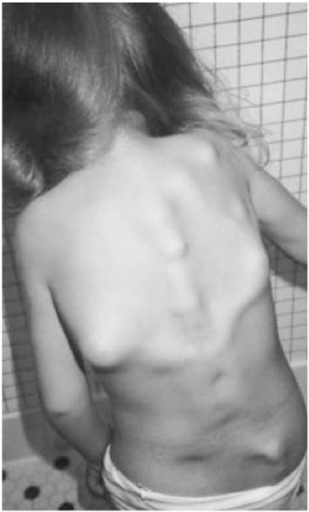
Abbildung 2: Schwellungen und Ausstülpungen, die für FOP charakteristisch sind. 5
Neue Therapien für FOP
Derzeit gibt es keine definitive Heilung für FOP. Allerdings haben jüngste Fortschritte bei zielgerichteten Therapien ein vielversprechendes Potenzial gezeigt, das Fortschreiten der Krankheit zu verlangsamen und die heterotope Ossifikation zu reduzieren.
- Palovarotene: ein selektiver Retinsäure-Rezeptor-Gamma (RARγ)-Agonist, ist das erste Medikament, das für die Behandlung von FOP zugelassen ist. Es wirkt, indem es die Knorpelbildung hemmt und so die Entwicklung von heterotopem Knochen reduziert. Es ist in Kanada zugelassen und wird in verschiedenen Ländern, darunter den USA und Europa, behördlich geprüft.
- Garetosmab: ein monoklonaler Antikörper, der auf Activin A abzielt, ein Protein, das an dem anomalen Signalweg beteiligt ist, der für die heterotope Ossifikation bei FOP verantwortlich ist.
- ACVR1-Inhibitoren: Da der primäre genetische Treiber von FOP eine Funktionsgewinnmutation im ACVR1-Gen ist, konzentrieren sich die Bemühungen auf die Entwicklung von Inhibitoren, die auf diesen Signalweg abzielen. Derzeit werden niedermolekulare Inhibitoren untersucht, die darauf abzielen, eine übermäßige Signalisierung des knochenmorphogenetischen Proteins (BMP) zu unterdrücken und HO zu verhindern.
- Gentherapie: In der Forschung werden CRISPR-basierte oder RNA-gerichtete Therapien zur Korrektur der ACVR1-Mutation untersucht.
- Entzündungshemmende Strategien: Einige Studien deuten darauf hin, dass eine frühzeitige entzündungshemmende Intervention bei Schüben die Entwicklung von HO abschwächen kann.
Es sind noch viele Schritte zu gehen
Obwohl es keine Heilbehandlung gibt, markieren laufende klinische Studien und behördliche Zulassungen einen bedeutenden Fortschritt im Umgang mit FOP. Die fortgesetzte Erforschung gezielter Therapien gibt Hoffnung, das Fortschreiten der Krankheit zu verlangsamen und die Lebensqualität der Betroffenen zu verbessern. FOP ist eine sehr seltene Krankheit, über deren Natur und Verlauf wir noch viel zu wenig wissen.