- Scarpa M. et al. (2023). Acid sphingomyelinase deficiency (ASMD): addressing knowledge gaps in unmet needs and patient journey in Italy-a Delphi consensus. Intern Emerg Med. 2023 Apr;18(3):831-842. doi: 10.1007/s11739-023-03238-3. Epub 2023 Mar 7. PMID: 36882619.
- McGovern MM. et al. (2017). Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. 2017 Feb 23;12(1):41. doi: 10.1186/s13023-017-0572-x. PMID: 28228103; PMCID: PMC5322625.
- McGovern MM. et al. (2004). Lipid abnormalities in children with types A and B Niemann Pick disease. J Pediatr. 2004;145:77–81. doi: 10.1016/j.jpeds.2004.02.048.
- McGovern MM. et al. (2017). Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med. 2017 Sep;19(9):967-974. doi: 10.1038/gim.2017.7. Epub 2017 Apr 13. PMID: 28406489; PMCID: PMC5589980.
ASMD: eine Krankheit, die immer wieder aufs Neue überrascht
Die ASMD ist eine äußerst seltene, jedoch potenziell lebensbedrohliche Erkrankung. Der lysosomalen autosomal-rezessiv vererbten Speicherkrankheit liegen SMPD1-Mutationen zugrunde. Was sind die wichtigsten Differentialdiagnosen?
ASMD: Aktivitätsmangel der sauren Sphingomyelinase
Aktuell bestehen diverse Wissenslücken zur ASMD (Mangel an saurer Sphingomyelinase). Es wird zwischen 3 Phänotypen unterschieden: der Niemann-Pick-Krankheit (NPD) Typ A (NPD A), Typ B (NPD B) und dem intermediären Typ A/B. Typ A und B der ASMD zählten früher gemeinsam mit dem Typ C zum Morbus Niemann-Pick. Heutzutage ist jedoch bekannt, dass dem Typ C nicht etwa ein Mangel an saurer Sphingomyelinase, sondern in 95 % der Fälle eine Mutation des NP-C1-Gens zugrunde liegt. Durch den beeinträchtigten Lipidtransport kommt es beim Typ C zur anormalen Cholesterinspeicherung. Typ A und B hingegen zählen zu den Sphingolipidosen.1-3
Bei den verschiedenen Typen der ASMD liegt ein Aktivitätsmangel der sauren Sphingomyelinase vor. Hierdurch kommt es zur Ansammlung von Sphingomyelin in unterschiedlichen Geweben des Körpers (Leber, Milz, Lunge, Knochen und ZNS). Die Patienten leiden unter einer Hepatosplenomegalie, einer interstitiellen Lungenerkrankung, Herzerkrankungen, Skelettanomalien, Wachstumsverzögerungen und hämatologischen Anomalien. Sie weisen ein atherogenes Lipidprofil auf.1,2
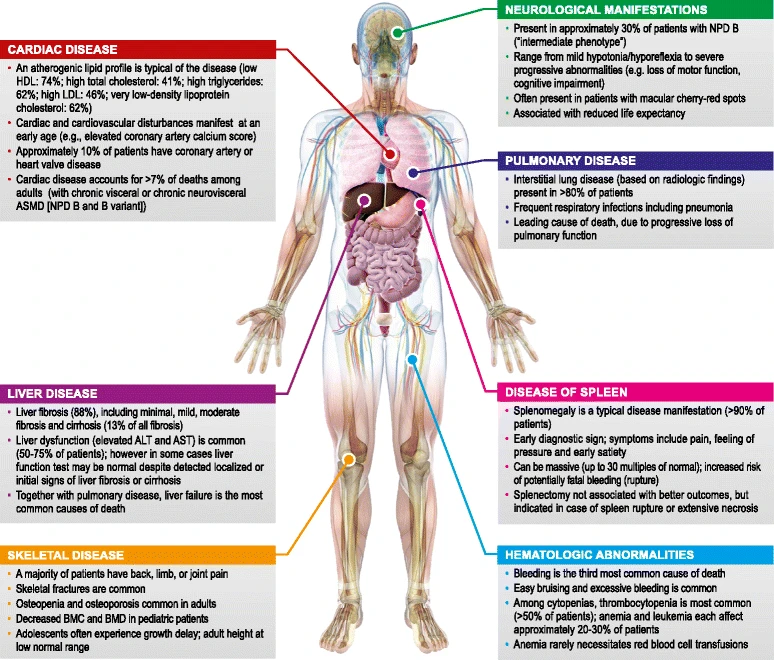
Abbildung 1: Im Vergleich zum Typ A weisen Patienten mit NPD B eine große phänotypische Heterogenität auf. In der vorliegenden Abbildung zeigen McGovern MM. et al. die unterschiedlichen ASMD-Manifestationen bei Patienten mit NPD B auf. [ALT: Alanin-Aminotransferasen; ASMD: Mangel an saurer Sphingomyelinase; AST: Aspartat-Aminotransferase; BMC: Knochenmineralgehalt; BMD: Knochenmineraldichte; NPD: Niemann-Pick-Krankheit.]2
Die Hälfte der ASMD-Patienten leidet unter hämatologischen Anomalien
In einer Querschnittsstudie von McGovern MM. et al. aus dem Jahr 2004 lagen bei fast der Hälfte der Patienten mit NPD A und B hämatologische Anomalien vor. 49 % der Patienten litten unter Blutungsepisoden, wobei das häufigste Blutungsereignis eine rezidivierende Epistaxis (29 % der Patienten) war. Folgende signifikante Blutungsereignisse traten vereinzelt in der besagten Querschnittsstudie auf:
- übermäßige Blutungen nach Tonsillektomie oder Adenoidektomie mit der Notwendigkeit einer Bluttransfusion
- Menorrhagie und Uterusblutungen mit der Notwendigkeit einer Hysterektomie
- Subduralhämatom
- Hämatemesis
- Hämoptyse
- Hämothorax
Bei 53 % der Patienten lag eine Thrombozytopenie vor. 26 % der Patienten litten unter Anämie und 21 % unter Leukopenie.2,3
Die wichtigsten Differentialdiagnosen der ASMD
Zu den Differentialdiagnosen der ASMD gehören u.a. andere Speicherkrankheiten wie die lysosomale saure Lipase-Defizienz (LAL-D), Morbus Gaucher und Niemann-Pick Typ C. Der Großteil der ASMD-Patienten leidet unter einer Hepatosplenomegalie mit erhöhten Transaminasen. Im Verlauf kann es zum fibrotischen Umbau der Leber und der Entwicklung einer Leberzirrhose kommen. Zu den wichtigsten Differentialdiagnosen hinsichtlich der Hepatosplenomegalie gehören primäre Lebererkrankungen (z. B. kryptogene Zirrhose, chronische Hepatitis B, Fettleber sowie autoimmun bedingte Leberveränderungen) sowie bösartige Erkrankungen. Im Vergleich zu anderen Stoffwechselkrankheiten wie der Mevalonatkinase-Mangel, der lysinurischen Proteinintoleranz und dem Transaldolase-Mangel ist die Hepatosplenomegalie bei ASMD deutlich stärker ausgeprägt. Auch bei der Farber-Krankheit, den Mukopolysaccharidosen und den GM1- und GM2-Gangliosidosen fällt die Hepatosplenomegalie milder aus. Bei der ASMD zeigt sich ein typisches Lipidprofil, das durch erhöhte LDL-Cholesterin-, VLDL-Cholesterin- und Triglyceridwerte gekennzeichnet ist. Die HDL-Cholesterinwerte sind deutlich vermindert. Ein ähnliches Lipidprofil ist bei LALD zu beobachten. Auch bei Patienten mit Gaucher-Krankheit liegt niedrige HDL-Cholesterinwerte vor – im Vergleich zur ASMD sind diese jedoch höher.1-3