Therapie und Verlauf:
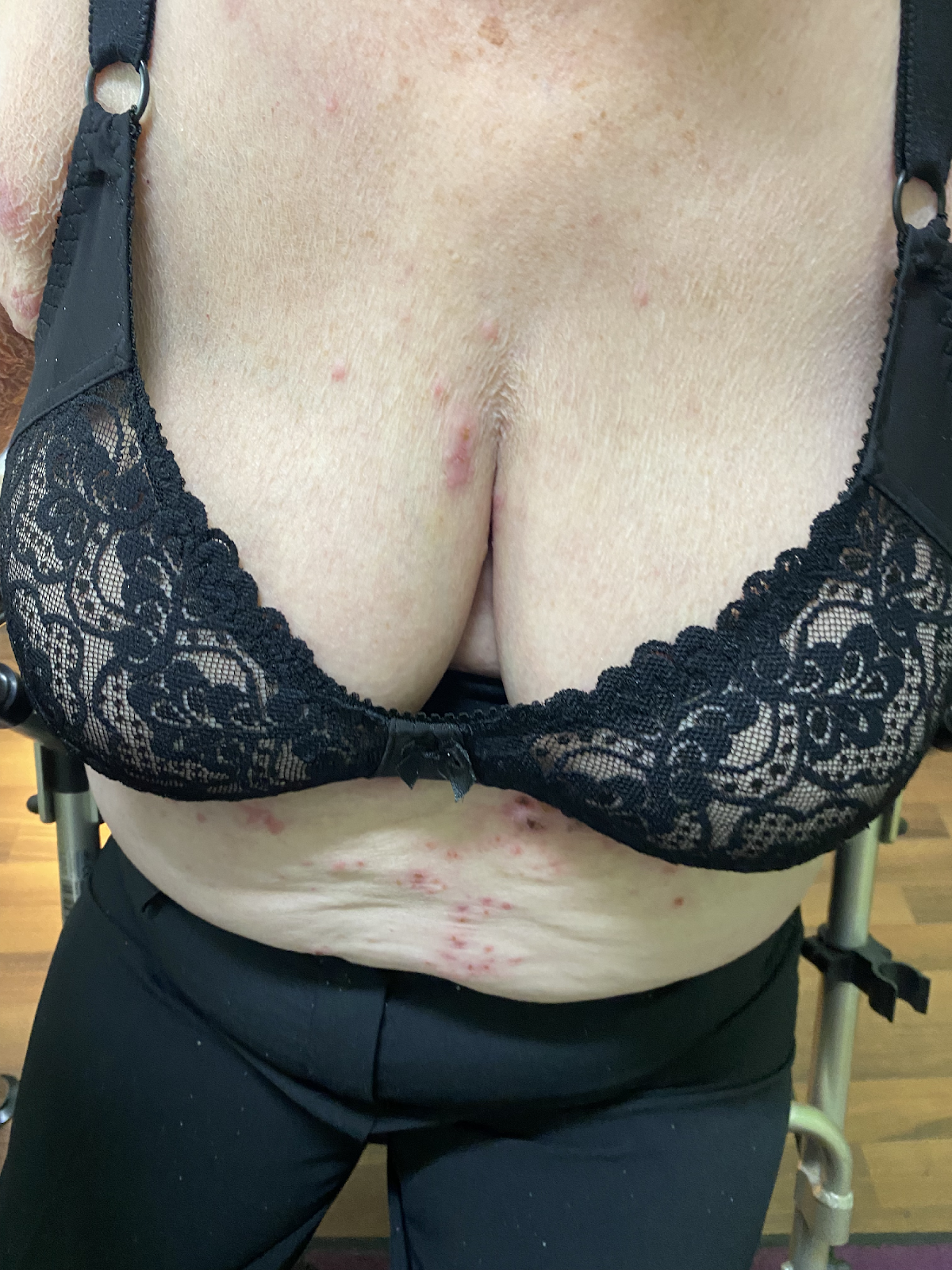
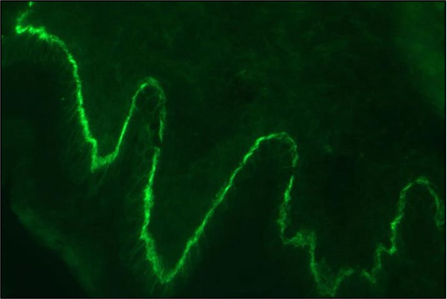
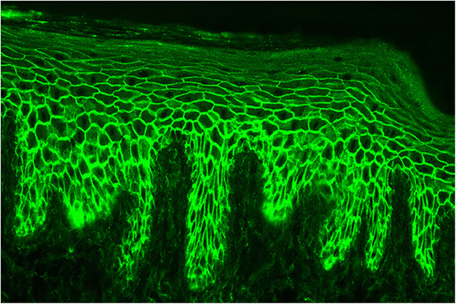
Die großen, stabilen serösen Spannungsblasen sowie die anamnestisch bestätigten, bereits länger bestehenden Hautläsionen am unteren Rumpf und den proximalen Extremitäten bei einer älteren Patientin deuteten schon vor Vorliegen der paraklinischen und bioptischen Diagnostikergebnisse stark auf ein bullöses Pemphigoid (BP) hin. Rückblickend charakteristisch sind die der Blaseneruption vorausgegangenen Hautläsionen, die zumeist ekzematös und urtikariell anmuten und als "prämonitorisches Erythem“ der Blasenbildung Monate bis Jahre vorangehen können. Die Verdachtsdiagnose BP wurde autoimmunserologisch, histologisch und durch die direkte Immunfluoreszenz bestätigt.
Schon vor Erhalt der Befunde wurde eine systemische steroidale Therapie mit Prednisolon 1mg/kgKG eingeleitet. Lokal wurde zusätzlich Betamethason-Creme appliziert. Die Juckreizstillung erfolgte mit Levocetirizin p.o. und Doxepin p.o.. Unter der Behandlung kam es zur relativ raschen Rückbildung der akuten Hautläsionen und zum Stoppen neuer Blasenbildung. Über einen Zeitraum von 14 Tagen konnte die Steroiddosis auf 10mg/d p.o. reduziert werden. Außerdem wurde die antidiabetische Behandlung mit Sitagliptin beendet und durch ein zusätzliches langwirksames Insulin ersetzt.
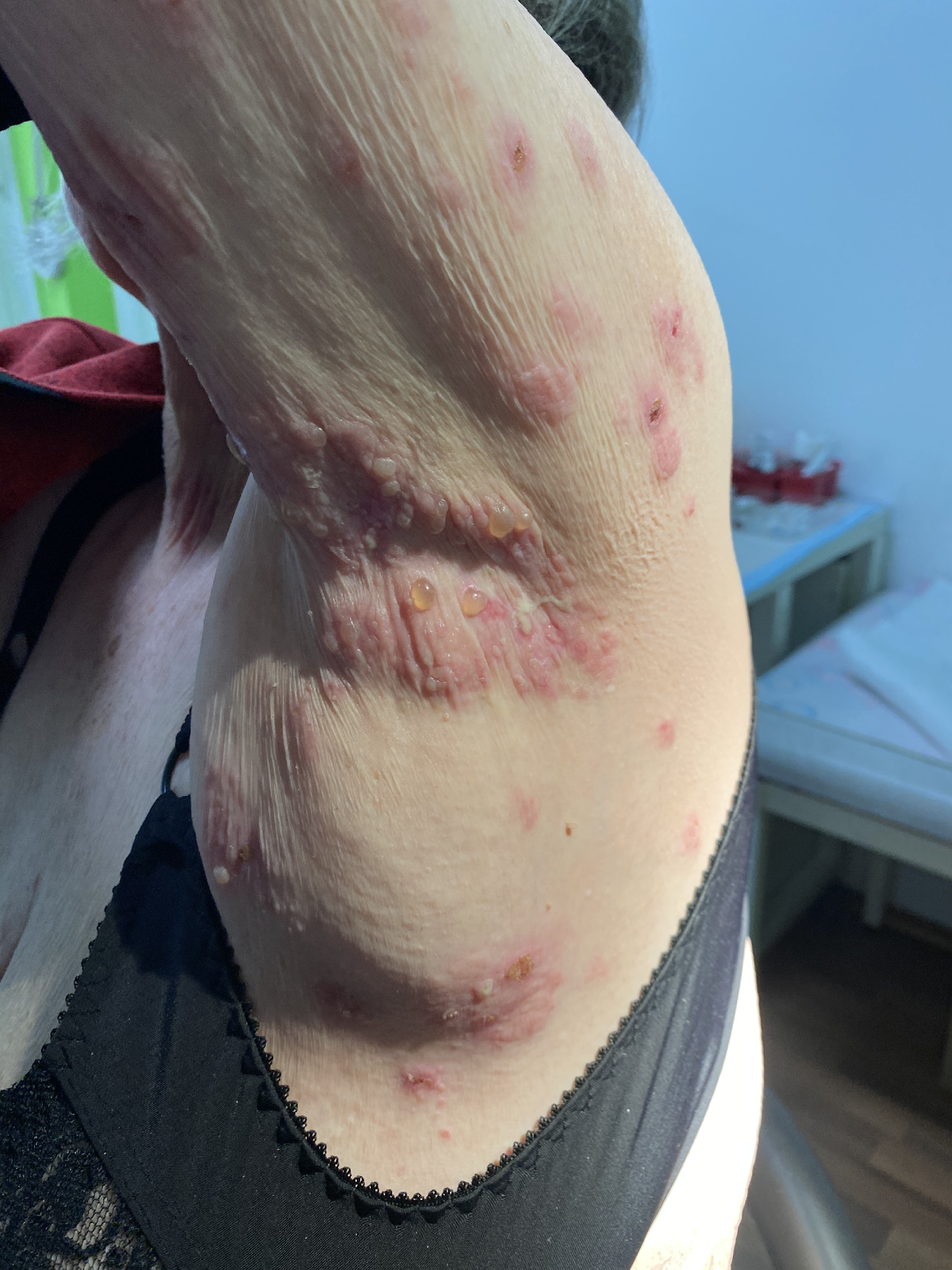
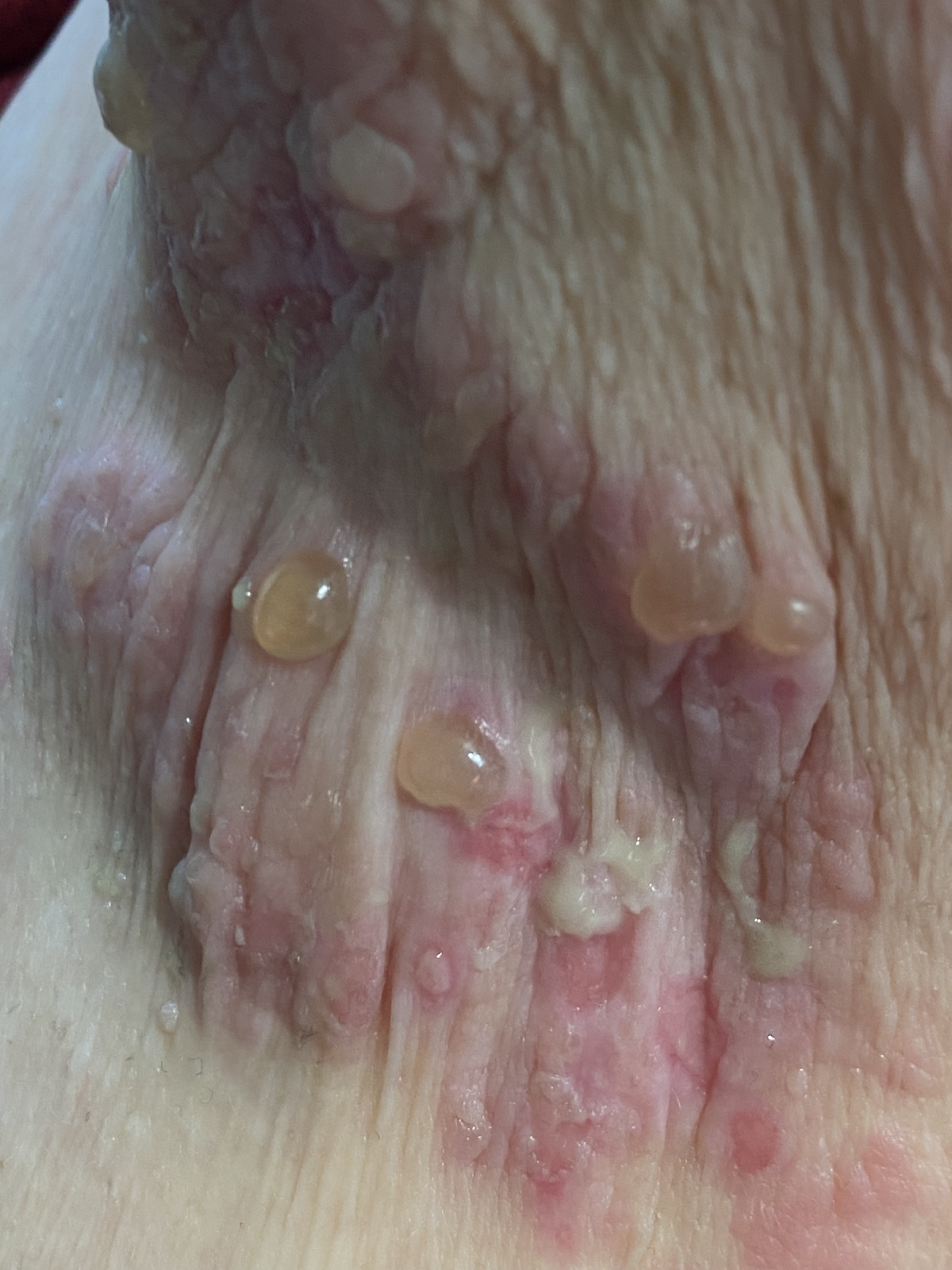
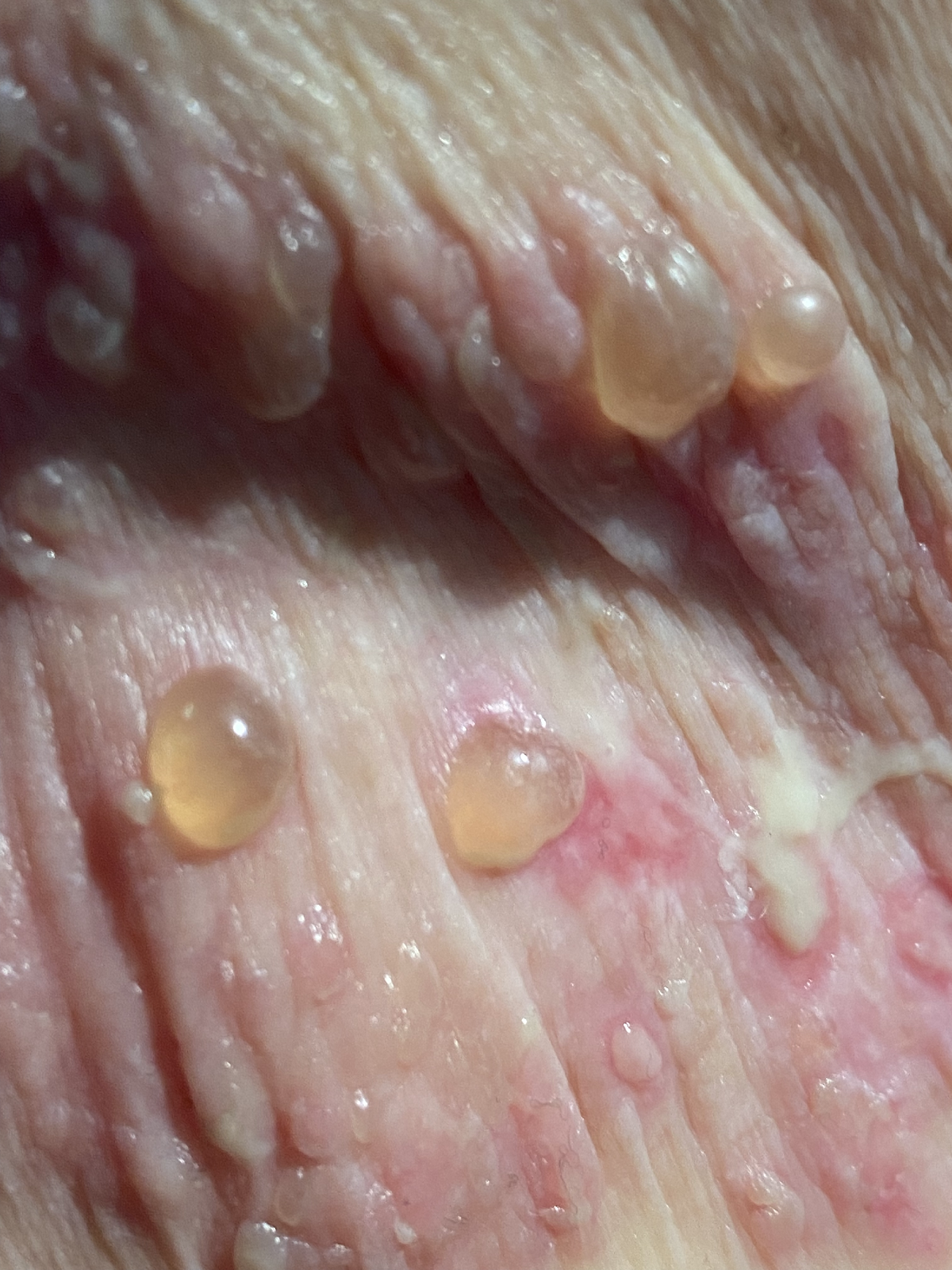
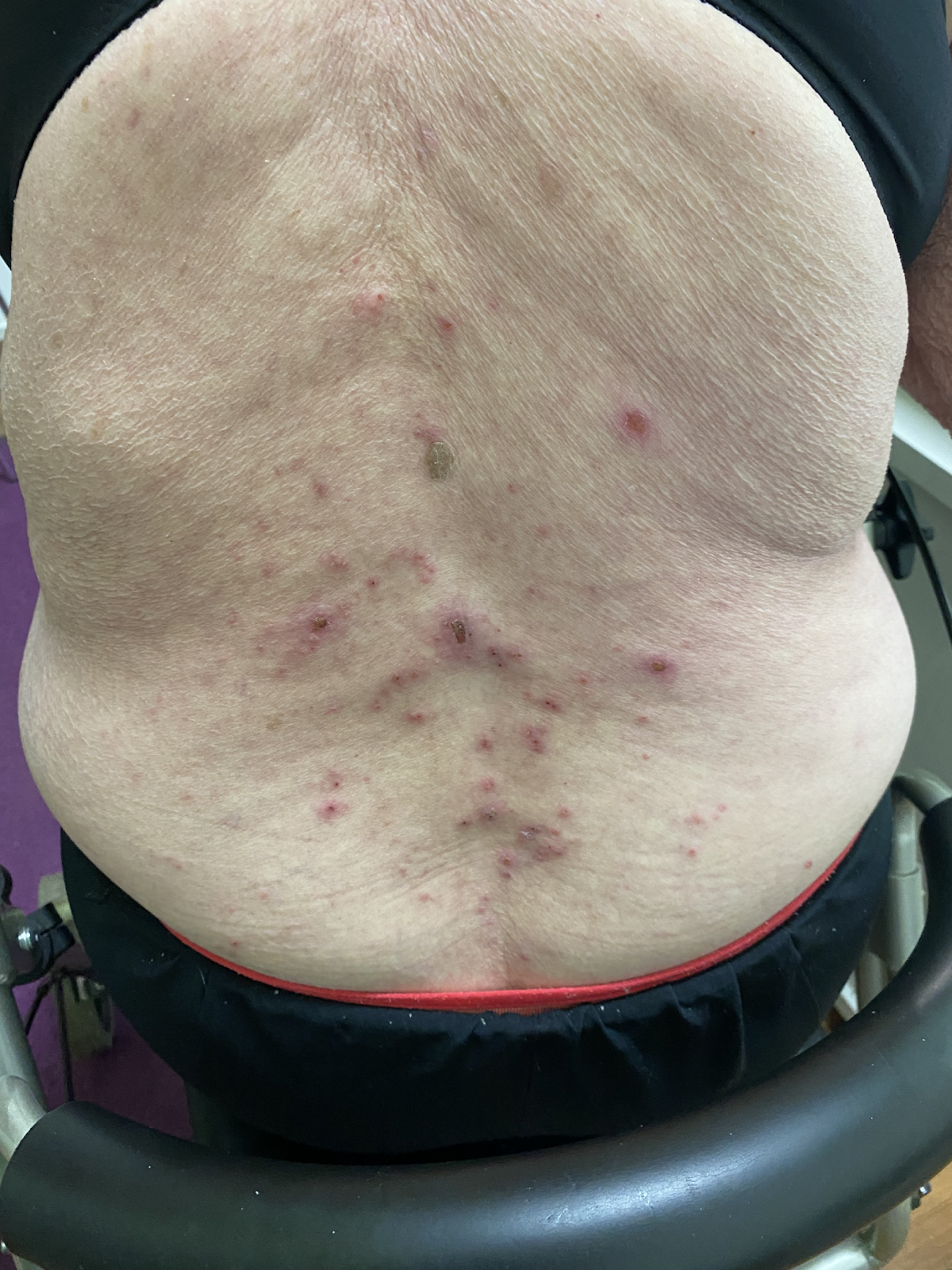
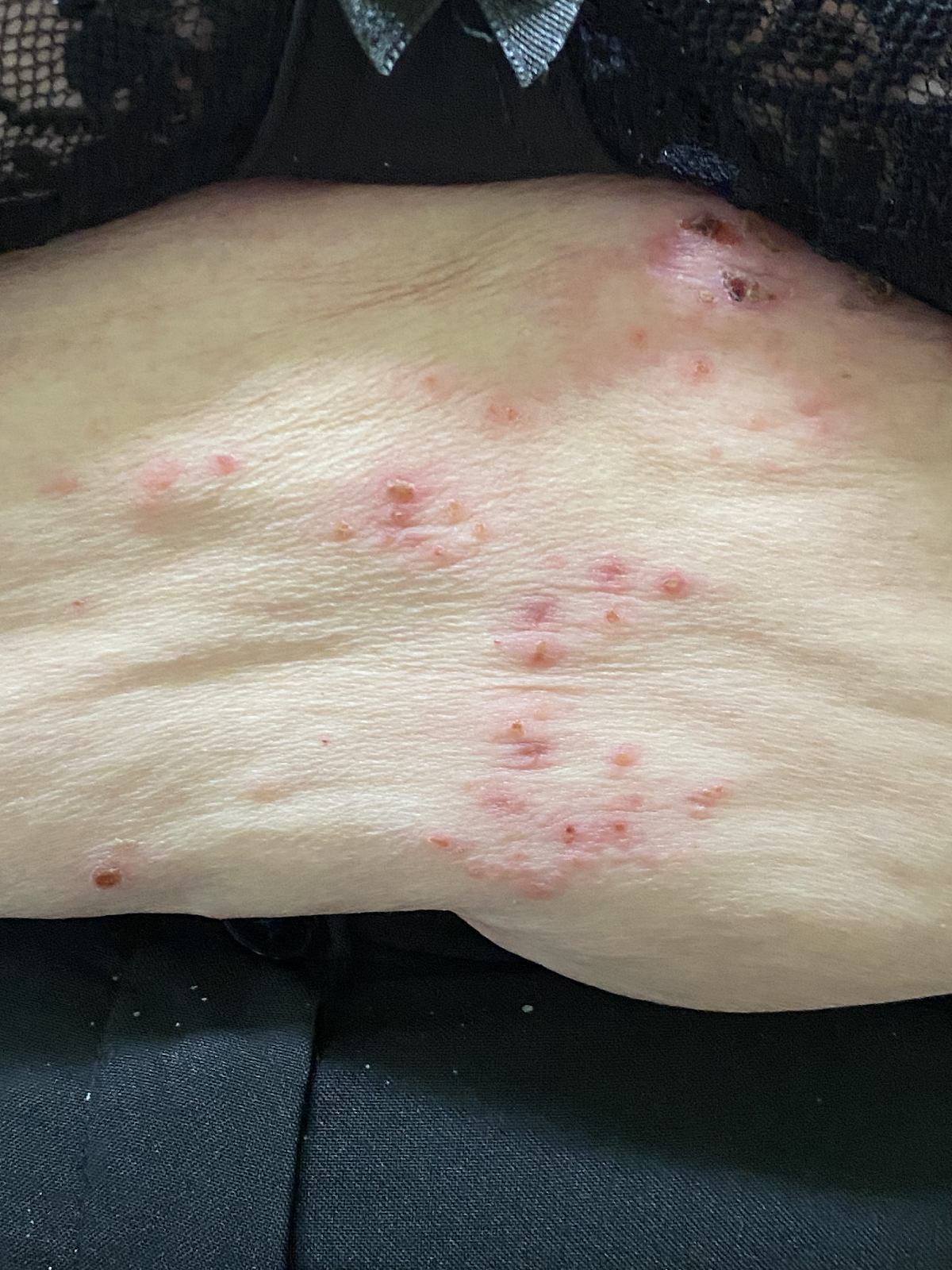
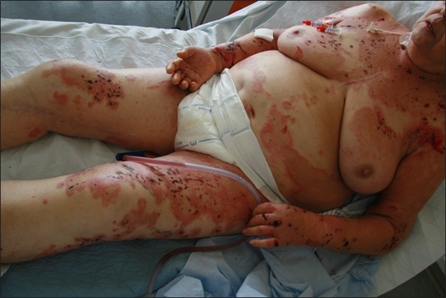
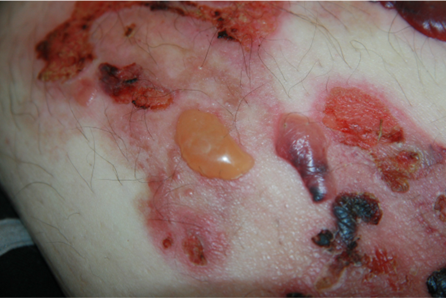
Das bullöse Pemphigoid ist mit einer Inzidenz von 10-34/ 1 Millionen Einwohner/Jahr die häufigste blasenbildende Immundermatose in Europa.1 Die meisten Patienten sind älter als 60 Jahre. Autoantikörperkomplexe, die sich gegen hemidesmosomale Strukturproteine der Basalmembran richten, BP180 (BPAg2) und BP230 (BpAg1), bewirken über Komplementaktivierung und Leukozytenmigration deren Ablösung. Damit stellt die gesamte Epidermis das Blasendach dar und die Folge sind sehr stabile Blasen, sogenannte Spannungsblasen. Die Blasen sind zumeist seriös, insbesondere aber bei Blutungsneigung, etwa bei der Einnahme von oralen Antikoagulanzien (OAK) können diese auch hämorrhagisch verändert sein (Abbildung 3).
a
b
Abb. 3: 75-jährige Patientin mit großflächig manifestiertem bullösem Pemphigoid, hier wird neben den teils hämorrhagischen Spannungsblasen auch die der Blasenbildung vorausgehenden teils ekzematös teils urtikarielle Hautläsionen sichtbar (a,b).
Seltener, in etwa 20% der Fälle, ist die Schleimhaut vor allem enoral beteiligt. Hier muss jedoch das vernarbende Schleimhautpemphigoid als eigene Entität unterschieden werden, welches ein heterogenes AK-Muster aufweist. Hierbei ist die Schleimhautbeteiligung führend und die vor allem konjunktivale Beteiligung führt die eigne Entität zu irreversibler Synblepharon- und Ektropiumbildung.
Die blasenbildende Autoimmunerkrankungen BAID stellen eine Gruppe von etwa 20 heterogenen Krankheitsbildern dar, die durch Autoantikörper gegenüber Strukturproteinen der Haut und/oder der Schleimhaut charakterisiert sind. Je nach Angriffsort der Autoantikörper werden subepidermale BAID (z.B. BP) von intraepidermalen BAID (z.B. Pemphigus vulgaris) unterschieden. Das bullöse Pemphigoid kann als Paraneoplasie auftreten (15%), wenngleich ein relevanter Malignomzusammenhang in früher Jahren eher überschätzt wurde.2 Die meisten Fälle sind idiopathischer Natur. Allerdings könne auch Medikamente gelegentlich das BP auslösen, z.B. Penicillin, Salazosulfasalazin, Diazepam oder Furosemid.
Auch ein Zusammenhang mit der Einnahme anderer Medikamente wie Spironolacton, Phenothiazinen oder Immun-Checkpoint-Inhibitoren wird diskutiert.3 In letzter Zeit aber häufen sich Fälle, bei denen das bullöse Pemphigoid durch Dipeptidylpeptidase-4 (DPP-4)-Inhibitoren Hemmer wie Sitagliptin getriggert wird.4 Pharmakovigilanzdaten weisen in der Tat in einem Teil der Fälle auf einen Zusammenhang mit der Einnahme von DPP-4-Inhibitoren hin, sodass empfohlen wird, diese bei Diagnose eines BP auf andere Antidiabetika umzustellen. Studiendaten sehen diesen Zusammenhang vor allem für männlichen Patienten.5
Im vorliegenden Fall ließ der zeitliche Zusammenhang mit der Einleitung der Sitagliptin-Medikation und dem Beginn der Hautsymptome vermuten, dass der DPP-4-Hemmer ursächlich war. Tatsächlich konnte nach dem Absetzen des Sitagliptin unter einer längeren Prednisolon-Erhaltungsdosis von 5mg/d ein dauerhaftes Abheilen der Autoimmundermatose erzielt werden.
Therapeutisch ist bei milden oder moderaten Verläufen in den nationalen Leitlinien zunächst eine gestaffelte Behandlung mit topischem Clobetasol und mit Prednisolon 0,5-1mg/kgKG empfohlen, bei rezidivierenden oder persistierenden Befunden werden zur Langzeitbehandlung Azathioprin, Dapson, Mycophenolatmoxetil oder Cyclophosphamid eingesetzt. Bei Schleimhautbefall, insbesondere konjunktival, wird topisch auch der Calcineurininhibitor Tacrolimus eingesetzt. Bei sehr therapierefraktären Verläufen kommen auch TNF-alpha-Inhibitoren oder der CD20-AK Rituximab zur Anwendung.6,7 Die europäische S2k-Leitlinie empfiehlt im Gegensatz zur deutschen Leitlinie den Einsatz von Rituximab bereits bei milden Verlaufsformen. Zudem wird zur Erhaltungstherapie die wiederholte Gabe von Rituximab nach 6, 12 und 18 Monaten angeraten.8
Differenzialdiagnostisch ist bei prämonitorischen BP vor allem an Prurigo, Urtikaria, Skabies aber lymphoproliferative Erkrankungen zu denken. Bei lokalisierter Blasenbildung sind mechanische oder traumatische Ursachen, Impetigo contagiosa, ein bullöses Erysipel, Herpes Zoster, bullöse Kontaktdermatitis, (bullöse) Iktusreaktion, generalisierte bullöse Arzneimittelreaktion und natürlich andere bullöse Autoimmundermatosen abzugrenzen.2
Fazit: Bullöses Pemphigoid effektiv mit modernen Therapien behandeln
Das bullöse Pemphigoid ist die häufigste Autoimmundermatose im höheren Lebensalter. Charakteristisch sind die stabilen Spannungsblasen am Rumpf und den proximalen Extremitäten. Der zumeist protrahierte Beginn als „prämonitorisches Erythem“ erklärt die oft späte Diagnosefindung. Das bullöse Pemphigoid ist zumeist idiopathisch, kann aber auch paraneoplastisch oder durch Arzneimittel getriggert auftreten. Hier häufen sich Beobachtungen im Zusammenhang mit der Gabe von DPP-4-Antagonisten. Die Therapie der Wahl im akuten Schub ist der systemische Steroidstoß, bei chronischem Verlauf sind klassische Immunsuppressiva wie Azathioprin und Cycloiphosphamid, aber auch moderne Biologika wie Rituximab empfohlen. Bei der Diagnose des bullösen Pemphigoids gilt es eine breite Differenzialdiagnose abzugrenzen.
- Deotto ML, Spiller A, Sernicola A et al. Bullous pemphigoid: An immune disorder related to aging (Review). Exp Ther Med 2022; 23: 50. Persson MSM, Begum N, Grainge MJ et al. The global incidence of bullous pemphigoid: a systematic review and meta-analysis. Br J Dermatol 2021
- Czaika et al: Kurzlehrbuch Dermatologie. Autoimmunerkrankungen der Haut. Thieme-Verlag, 3.Auf. 2ß23, S161ff.
- 14 Geisler AN, Phillips GS, Barrios DM et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol 2020; 83: 1255–1268. doi:10.1016/j.jaad.2020.03.132
- Mohme S et al. Blasenbildende Autoimmundermatosen - Klinik, Diagnostik und neue Therapieansätze. Akt Rheumatol 2022; 47: 333–343 © 2022. Thieme
- Lee SG et al. Association of Dipeptidyl Peptidase 4 Inhibitor Use With Risk of Bullous Pemphigoid in Patients With Diabetes. JAMA Dermatol 2019;155(2):172-177
- Schmidt E, Goebeler M, Hertl M et al. S2k guideline for the diagnosis of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 2015; 13: 713–727.
- Chen DM, Odueyungbo A, Csinady E et al. Rituximab is an effective treatment in patients with pemphigus vulgaris and demonstrates a steroid-sparing effect. Br J Dermatol 2020; 182: 1111–1119
- Joly P, Horvath B, Patsatsi Α et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol 2020; 34: 1900–1913